MedDeviceGuide — IVD & Medical Device Knowledge Base

India's Medical Device Market Decoded: 4,108 Manufacturers, 62% Imports
India's medical device market reached $20.98B in 2026 with 4,108 licensed manufacturers. Class A and B devices dominate at 76%, while imports supply 62% of market demand.

Colombia INVIMA: 4,582 Importers, 80% Imports — Who Controls Device Access?
Analysis of 423,394 INVIMA records reveals 4,582 importers and 2,537 apoderados competing in Colombia's USD 1.5B device market, with 81.8% of registrations tied to import pathways.

EU MDR Post-Market Surveillance Plan: MDCG 2025-10 Practical Guide
Build a PMS plan aligned with MDCG 2025-10 guidance: proactive data collection, QMS integration, trend reporting, PMS reports vs PSURs, and custom-made device obligations.

EU-Switzerland MRA Update 2026: What It Means for Medical Device Market Access
How the EU-Switzerland MRA signed March 2, 2026 restores medical device mutual recognition, what changes for EU and Swiss manufacturers, swissdamed timelines, and the path to ratification.

EUDAMED at Scale: 1.29 Million UDI-DIs Reveal Who Supplies the EU Device Market
Analysis of 1,292,737 EUDAMED UDI-DI records shows China leads with 19.1% of registrations, Turkey contributes 10.3% Class III devices, and four countries supply nearly half the EU market.

Indonesia's Device Registry: 78K Registrations, 76% Imported, Expiry Cliff
Analysis of Indonesia's Kemenkes device registry reveals 75.8% of registrations are imports (AKL), China supplies 36.5% of imported devices, and 17.2% of all registrations expire in 2026.

Medical Device Accessory Classification: EU MDR and FDA Pathways
How accessories are classified separately from parent devices under EU MDR Article 2(2) and FDA 513(f)(6), with examples, MDCG 2021-24 Rev.1 updates, and FDA's 2025-2026 reclassification initiative.

Aesthetic Device Regulation: FDA 510(k) and EU MDR for Laser, RF, and Energy Devices
How energy-based aesthetic devices — laser, RF, HIFU, IPL, cryolipolysis, and EMS — are classified and cleared by FDA and EU MDR, including special controls, Annex XVI, and 2026 compliance updates.

Asia-Pacific Medical Device Registration: Market Comparison and Entry Strategy (2026)
Compare medical device registration across APAC markets — China, Japan, South Korea, Australia, Singapore, India, ASEAN — including timelines, fees, reliance pathways, and strategic entry order.

IVD Calibrator Traceability: ISO 17511, EU IVDR, and JCTLM Guide
Metrological traceability for IVD calibrators and control materials under ISO 17511:2020 and EU IVDR Annex I Section 9.3, covering calibration hierarchies, JCTLM resources, and common pitfalls.

21 CFR Part 4: Combination Product cGMP Quality Systems for Drug-Device Manufacturers
How drug-device combination product manufacturers implement quality systems under 21 CFR Part 4 — streamlined approach, QMSR update, design controls, stability, and EU MDR Article 117.

FDA 510(k) Fees: MDUFA V User Fee Schedule and Total Cost Breakdown
Complete guide to FDA 510(k) user fees for FY 2026 under MDUFA V, including the $26,067 standard fee, $6,517 small business fee, $11,423 establishment registration, waivers, and total project cost.

ISO 19011:2026: What the Updated Audit Standard Means for Medical Device Manufacturers
ISO 19011:2026 embeds hybrid auditing, digital evidence management, and updated auditor competence across the audit lifecycle — what ISO 13485 and QMSR programs need to update.

Medical Device Post-Approval Change Management: FDA, EU MDR, and Asian Markets (2026)
When device modifications require new regulatory submissions in the US, EU, Korea, Japan, and China — FDA special 510(k), PMA supplements, EU MDR significant changes, and Asian pathways.

Veterinary Medical Device Regulation: FDA CVM, EU Gaps, and Global Strategy
How veterinary medical devices are regulated under FDA CVM, EU frameworks, and key global markets. Covers classification gaps, 510(k) exemptions, off-label use, and market entry strategy.

FDA Prior Knowledge Framework for Cell and Gene Therapy: Draft Guidance June 2026
FDA's June 2026 draft guidance on reusing CMC, nonclinical, and clinical prior knowledge to accelerate gene therapy development, and its interaction with the Plausible Mechanism Framework.

MedTech M&A Market Analysis H1 2026: Deal Trends, Strategic Shifts, and Outlook
Medical device M&A H1 2026: $26.6B Q1 deal value, 87% strategic buyers, Boston Scientific-Penumbra, Danaher-Masimo, portfolio reshaping trends, and H2 outlook for medtech dealmaking.

UK MHRA International Reliance Pathway: Draft Regulations 2026 Analysis
MHRA's draft Medical Devices Regulations 2026: International Reliance Pathway for US, Canada, and Australia approvals, IVD reclassification, PCCPs for AI software, and what manufacturers must prepare.
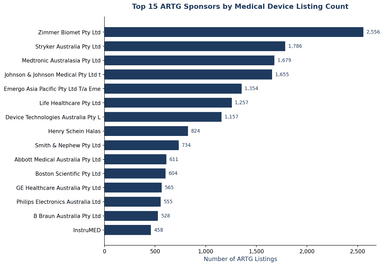
Australia ARTG Sponsor Map: Who Controls Market Access for 63,000 Devices?
Analysis of Australia's ARTG reveals 3,377 sponsors managing 63,131 device listings — top 10 sponsors control 22%, and Emergo Australia represents 375 overseas manufacturers.
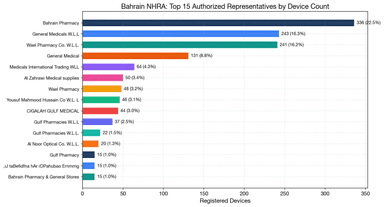
Bahrain NHRA: 3 ARs Hold 55% of Devices — Small GCC Market Channel Lock-In
Bahrain NHRA device database analysis: 62 ARs manage 1,491 devices, top 3 hold 55%, all tracked licenses expired, and manufacturer-AR lock-in is high. GCC channel strategy implications.